The study of complex microbial communities (metacommunities) and their interactions with the environment, with their hosts, or other organisms, involves mainly the analysis of their genetic potential (metagenomics), their activity in terms of expressed genes (metatranscriptomics) or the products of their activities (metaproteomics and metabolomics).
Description of microbial communities
A quick and cheap approach to the description of microbial communities consists of "labeling" the organisms present in a sample, to obtain their taxonomic annotation, associated with the count of the times they are found within the same sample, such as approximation of the relative abundance of each organism (taxon). This approach has been given various names such as meta-barcoding, gene-survey or, probably what we like best: "metataxonomy".
This name refers to the description of the taxonomy of a complex population or metapopulation. In general, it make use of universal primers to amplify the 16S ribosomal gene from - ideally - all the bacteria in a sample by means of PCR (Polymerase Chain Reaction). The term "metagenomics" is often misused for this type of approach. Actually, we use the term metagenomics to describe the entire genomic potential of a complex sample through an approach based on the random shearing of DNA extracted from a natural sample and its sequencing. In sensu-stricto metagenomics, there is no bias from selection imposed by primers chose or estimation errors derived from the polymerase chain reaction. We could say that what you see is what you get.
Metataxonomy
Amplicon-based microbial diversity descriptions focus on comparing 16S ribosomal gene amplicons and then using the counts in distribution analyses.

Metagenomics
In metagenomic analyses, the DNA coming from a sample containing a complex community is sheared randomly, sequenced, assembled and, finally, annotated. Functional and taxonomic profiles are then obtained and reported for downstream descriptive and statistic analysis.
In this article we are going to talk about the advances in the sequencing of 16S amplicons obtained with the new third-generation sequencing technologies, referring especially to the results of the new sequencer from the company Pacific Biosciences (PacBio) and its Sequel-II sequencer.
16S rDNA gene amplification
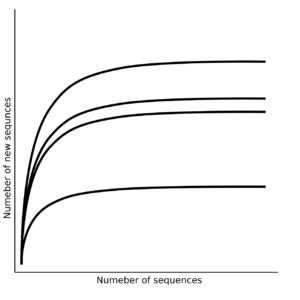
From the 1990s until the advent of second-generation methods, the description of taxa distributions in microbial communities was carried through a DNA extraction protocol, its amplification with universal primers, cloning of the amplicons in commercial plasmid vectors, transformation, selection of clones and individual sequencing of each insert from each clone using the famous (and still in use) Sanger method (first generation). Second-generation methods have made it possible to skip the cloning step and directly sequence the amplicons, albeit with previous steps of preparing sequencing libraries, producing, in the end hundreds of thousands of sequences in a single experiment. In this way, we have achieved a very high number of sequences per sample, and an unprecedented cost reduction per experiment. Descriptions of microbiota in the 1990s could be published with fewer than 100 sequences per sample, attempting to tell an ecological story with so little information. With the entry of the second generation of sequencers, the analyzes were carried out with such a high number of sequences per sample, that they allowed us to effortlessly saturate all the rarefaction curves, even from the most diverse environments.
Rarefaction
Rarefaction curves. The X axis represents the number of considered sequences for each sample (lines); the Y axis reports the number of observed unique sequences.
But what have we lost? We have lost in specificity. Second generation methods cannot sequence beyond 300 base pairs (even with specific protocols to sequence an amplicon by two directions to obtain a longer read extension). In general, studies on the distribution of the 16S ribosomal gene are carried out using amplicons with a size of around 450 base pairs (in the best of cases) and that include, at most, two hypervariable regions of said gene. This information is then used to identify the most likely taxonomic rank by matching each representative sequence against a database (as one of the most common approaches). The problem we face is that in order to obtain a reliable taxonomic assignment down to the rank of species (or clone) we need the sequence of the complete 16S ribosomal gene. This is impossible to achieve using second generation methods due to their technical limitation.
Even so, low cost, simplicity in data collection and development, bioinformatic tools, and lack of alternatives have made partial sequencing of the 16S ribosomal gene (about one third of the full gene length) the standard for microbial community characterization. As a result, we have been populating databases of partial sequences for about 10 years.
We do not have to forget that the phylogenetic evidence that led Carl Woese to describe the taxonomic importance of this gene is based on alignments that include all 9 hypervariable regions of this gene (Woese and Fox. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. PNAS USA. 1977 Nov; 74(11):5088-90). In the 1990s, even when technology and associated statistics were limiting factors, the cloning method associated with Sanger sequencing made it possible to obtain the complete 16S ribosomal gene (about 1540 base pairs) with a reliable taxonomic annotation up to the level of species. With the advent of second-generation methods, we are now used to and happy with partial sequence annotations that, in general, do not go beyond the genus level, mainly due to lack of genetic information.
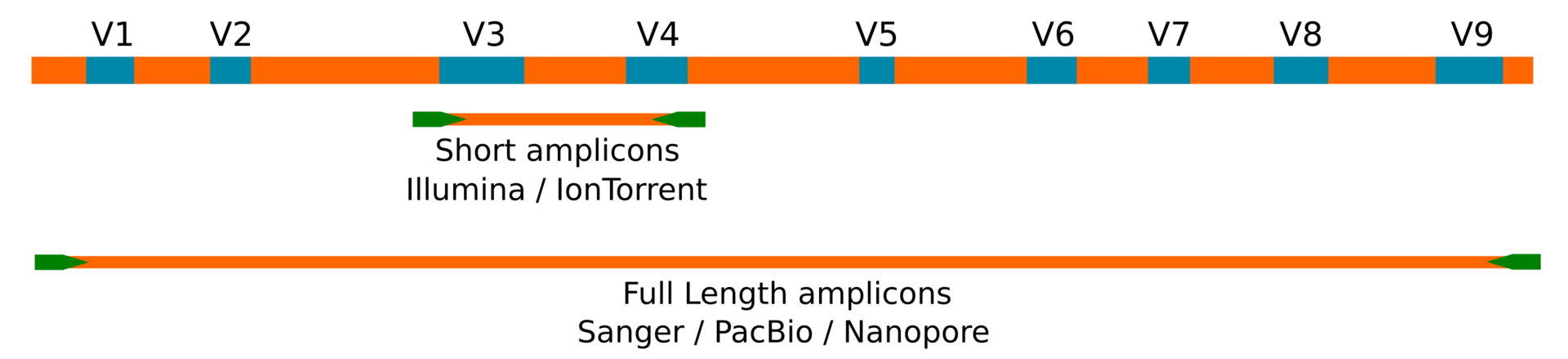
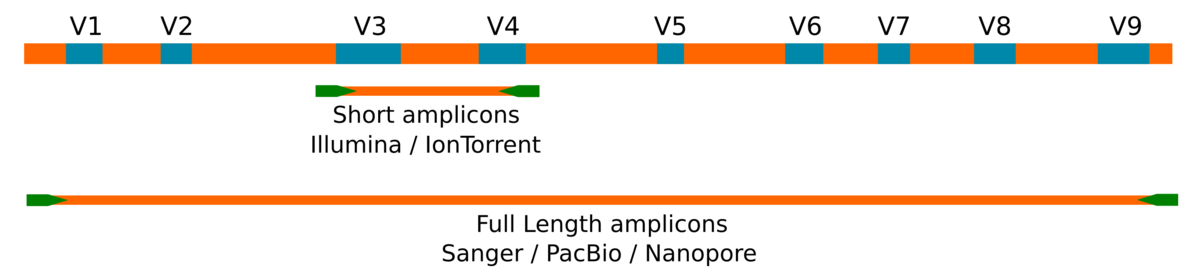
Amplicons
The second generation Illumina and IonTorrent methods produce sequences of about ~500bp. Sanger, PacBio and Nanopore, allow to reach the entire gene with paired or unique sequences of ~1500bp.
Even so, low cost, simplicity in data collection and development, bioinformatic tools, and lack of alternatives have made partial sequencing of the 16S ribosomal gene (about one third of the full gene length) the standard for microbial community characterization. As a result, we have been populating databases of partial sequences for about 10 years.
We do not have to forget that the phylogenetic evidence that led Carl Woese to describe the taxonomic importance of this gene is based on alignments that include all 9 hypervariable regions of this gene (Woese and Fox. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. PNAS USA. 1977 Nov; 74(11):5088-90). In the 1990s, even when technology and associated statistics were limiting factors, the cloning method associated with Sanger sequencing made it possible to obtain the complete 16S ribosomal gene (about 1540 base pairs) with a reliable taxonomic annotation up to the level of species. With the advent of second-generation methods, we are now used to and happy with partial sequence annotations that, in general, do not go beyond the genus level, mainly due to lack of genetic information.
Sequencing of the Full Length 16S ribosomal gene (FL-16S)
In 2009, third generation methods from Pacific Biosciences (PacBio) and Oxford Nanopore Technologies finally arrive. Both promised very long sequences. The doubts remain in the quality values of the readings. While Nanopore, to date, covers a significant number of applications due to the simplicity in the preparation of libraries and sequencing, the low cost of infrastructure, the speed in obtaining data and the portability, PacBio stands out for the quality of the data. Both methods produce sequences with lengths that can exceed one hundred kilobases (100,000 nucleotides), being nowadays, an obligatory choice in the sequencing of complete genomes. The low quality of Nanopore is usually corrected with coverage and/or hybrid sequencing (Nanopore + Illumina).
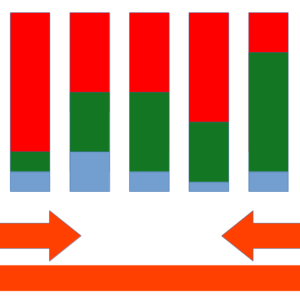
PacBio already offers its new HiFi sequencing protocol, with very high quality sequences, whose quality is averaging "Q50" with peaks of "Q90", that is, one error every 100,000-1,000,000,000 nucleotides. This makes it suitable for taxonomic description analysis where we need a high degree of confidence for a correct taxonomic annotation of each of the obtained sequences. What makes PacBio more interesting for these types of studies is that we can finally re-sequence the entire 16S ribosomal gene and annotate down to species (or clone) rank, using even the entire ribosomal operon, at a cost per sample very close to the one offered by the second generation sequencing methods. We have returned to the era of complete gene sequencing, but with the advantages of low-cost, high-quality data in an impressively short sequencing time, typical of modern parallel sequencing methods.
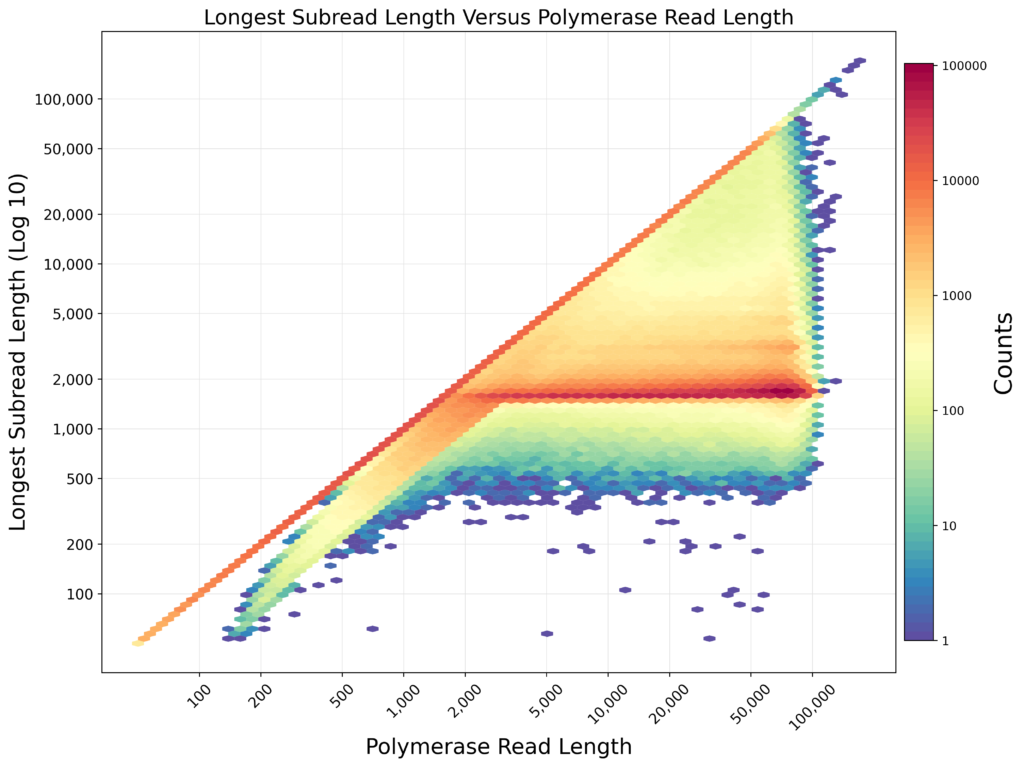
Representation of the quality of PacBio sequencing of 16S ribosomal gene amplicons. PacBio produces sequences that are read multiple times (X axis), improving the quality. The Subreads represent the multiple readings already collapsed into one (Y axis). The color map represents the number of sequences.
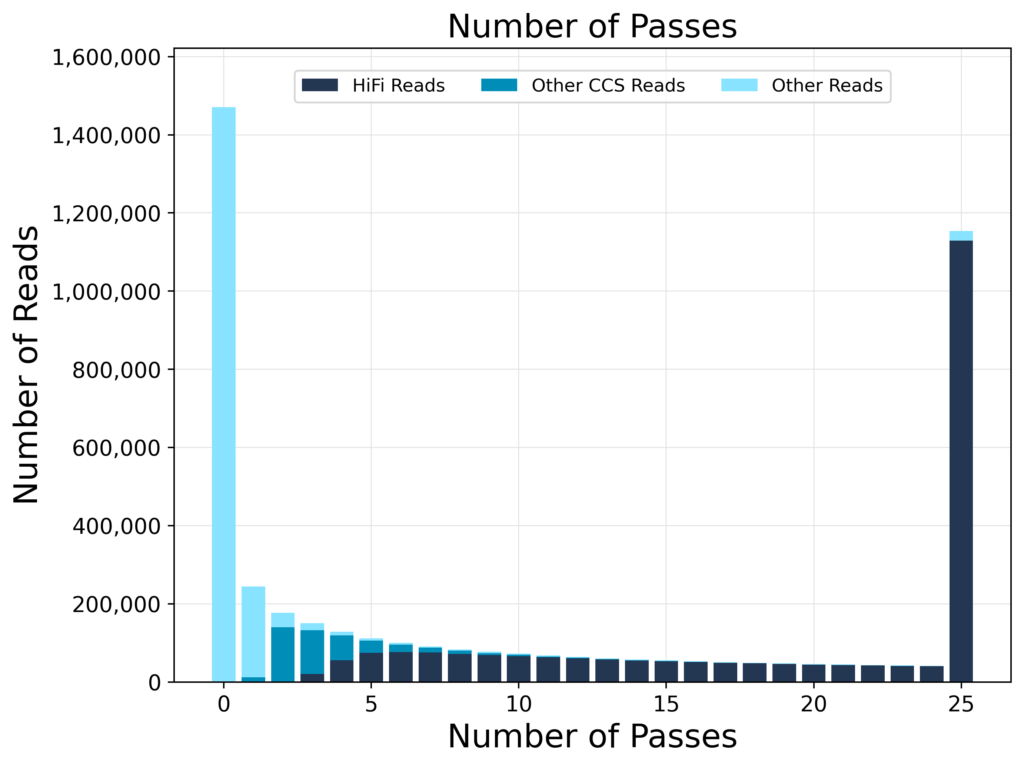
NumberOfPasses
Each PacBio sequence is read multiple times improving the quality. Those that exceed a threshold number of steps are defined "HiFi".
Now we are at the point where we have to "repopulate" the databases with the complete 16S ribosomal gene sequences. Paradoxically, even having the complete information in our files, we find ourselves with around ten years of partial data accumulated in the databases. Today, this "lack of information" represents a problem that, step by step, will be solved with the contribution of new, complete and reliable information.
More on PacBio applications in metagenomics at: PacBio BLOG: https://www.pacb.com/blog/cost-effective-metagenomics/