El estudio de las comunidades microbianas complejas (metacomunidades) y sus interacciones con el medio ambiente, con sus hospedadores, u otros organismos, pasa principalmente por el análisis de sus potencialidades genéticas (metagenómica), de su actividad en términos de genes expresados (metatranscriptómica) o de los productos de sus actividades (metaproteómica y metabolómica).
Descripción de las comunidades microbianas
Un primer paso, rápido y barato, para la descripción de comunidades microbianas consiste en el "etiquetado" de los organismos presentes en una muestra, para obtener su anotación taxonómica, asociada al conteo de las veces que lo encontramos dentro de la misma muestra, como aproximación de la abundancia relativa de cada organismo (taxón). Este abordaje ha recibido varios nombres como, por ejemplo, meta-barcoding, gene-survey o, probablemente lo que más nos gusta: "metataxonomy". Este nombre hace referencia a la descripción de la taxonomía de una población compleja o metapoblación. Por lo general se trata de usar cebadores universales para amplificar mediante PCR (Polymerase Chain Reaction) el gen ribosomal 16S de - idealmente - todas las bacterias de una muestra. A menudo el término de metagenómica se usa incorrectamente para este tipo de abordaje. En realidad, el término de metagenómica lo usamos para describir todo el potencial genómico de una muestra compleja mediante un abordaje basado en el corte al azar de DNA extraído de una muestra natural y su secuenciación. En metagenómica sensu-stricto, no hay ningún sesgo procedente de la selección impuesta por los cebadores o por los errores de estimaciones derivados de la reacción en cadena de la polimerasa. Podríamos decir que lo que ves es lo que hay (what you see is what you get).
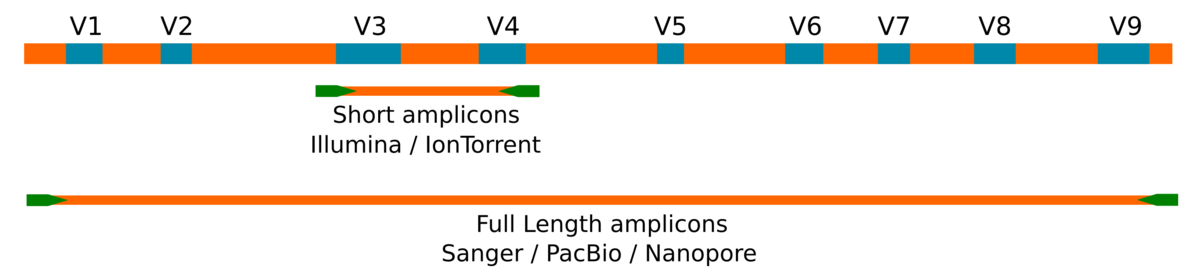
Los análisis de diversidad microbiana basados en amplicones se enfocan en la comparación de amplicones del gen ribosomal 16S para luego usar los recuentos en los análisis de distribuciones.

En los análisis metagenómicos, el ADN procedente de una muestra que contiene una comunidad compleja se corta al azar, se secuencia, se ensambla y, finalmente, se anota. Luego los perfiles funcionales y taxonómicos obtenidos se usan para el análisis descriptivo y estadístico posterior.
En este artículo vamos a hablar sobre los avances en la secuenciación de amplicones de 16S obtenidos con las nuevas tecnologías de secuenciación de tercera generación, refiriéndonos especialmente a los resultados del nuevo secuenciador de la compañía Pacific Bioscience (PacBio) y a su secuenciador Sequel-II.
Amplificación del gen ribosomal 16S
Desde los años noventa hasta la entrada de los métodos de segunda generación, la descripción de las distribuciones de taxones en comunidades microbianas se llevaba adelante mediante un tedioso protocolo de extracción de DNA, su amplificación con cebadores universales, clonación en vectores plasmídicos comerciales, transformación, selección de los clones y secuenciación individual de cada inserto de cada clon mediante el famoso (y todavía en uso) método de Sanger (primera generación). Los métodos de segunda generación han permitido saltar el paso de clonación y secuenciar directamente los amplicones, eso sí, con unos pasos previos de preparación de librerías de secuenciación, que al final producían en un único experimento, cientos de miles de secuencias. De esta forma hemos alcanzado una cantidad muy elevada de secuencias por muestra, y una reducción de costos por experimento, sin precedentes. Las descripciones de las microbiotas en los años 90's podían publicarse con menos de 100 secuencias por muestra, intentando describir una historia ecológica con tan poca información. Con la entrada de la segunda generación de secuenciadores, los análisis se llevaron a cabo con un número tan alto de secuencias por muestra, que nos permitieron saturar sin esfuerzo todas las curvas de rarefacción, hasta de los ambientes más diversos.
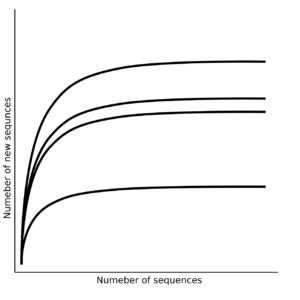
Curvas de rarefacción. El eje X representa el número de secuencias consideradas para cada muestra (líneas); el eje Y representa el número de secuencias únicas observadas.
¿Pero, que hemos perdido? Hemos perdido en especificidad. Los métodos de segunda generación no pueden secuenciar más allá de 300 pares de bases (aún con protocolos específicos para secuenciar un amplicon por dos direcciones para obtener una lectura más larga). En general los estudios de distribuciones del gen ribosomal 16S se llevan a cabo usando amplicones de un tamaño de alrededor de 450 pares de bases (en el mejor de los casos) y que incluyen, como mucho, dos regiones hipervariables de dicho gen. Esta información se usa luego para identificar el rango taxonómico más probable, alineando (u con otros métodos de búsqueda) contra una base de datos. El problema a lo que nos enfrentamos, es que para obtener una asignación taxonómica fiable hasta el rango de especie (o clon), se tendría que disponer del gen ribosomal 16S completo, imposible con los métodos de segunda generación por su limitación técnica, a excepción de algunos protocolos que usan métodos de concatenación de secuencias, que no son muy usados por sus costes y peculiaridades.
Aun así, el bajo coste, la simplicidad en la obtención de datos y el desarrollo, las herramientas bioinformáticas, y la falta de alternativas, han hecho que la secuenciación parcial del gen ribosomal 16S se haya convertido en el estándar en la caracterización de las comunidades microbianas. Como resultado, llevamos alrededor de 10 años rellenando las bases de datos de secuencias parciales.
No olvidemos que las evidencias filogenéticas que llevaron a Carl Woese a describir la importancia taxonómica de este gen, se basan en los alineamientos que incluyen todas las 9 regiones hipervariables de este gen (Woese and Fox. Phylogenetic structure of the prokaryotic domain: the primary kingdoms. PNAS USA. 1977 Nov; 74(11):5088-90 ). En los años 90's, aun cuando la tecnología y la estadística asociada eran factores limitantes, el método de clonación asociado a la secuenciación de Sanger permitía obtener el gen ribosomal 16S completo (alrededor de 1540 pares de bases) con una anotación taxonómica fiable hasta el nivel de especie (o clon). Con la llegada de los métodos de segunda generación ahora nos encontramos acostumbrados y contentos con anotaciones de secuencias parciales que, en general, no superan el nivel de género, principalmente por falta de información genética.
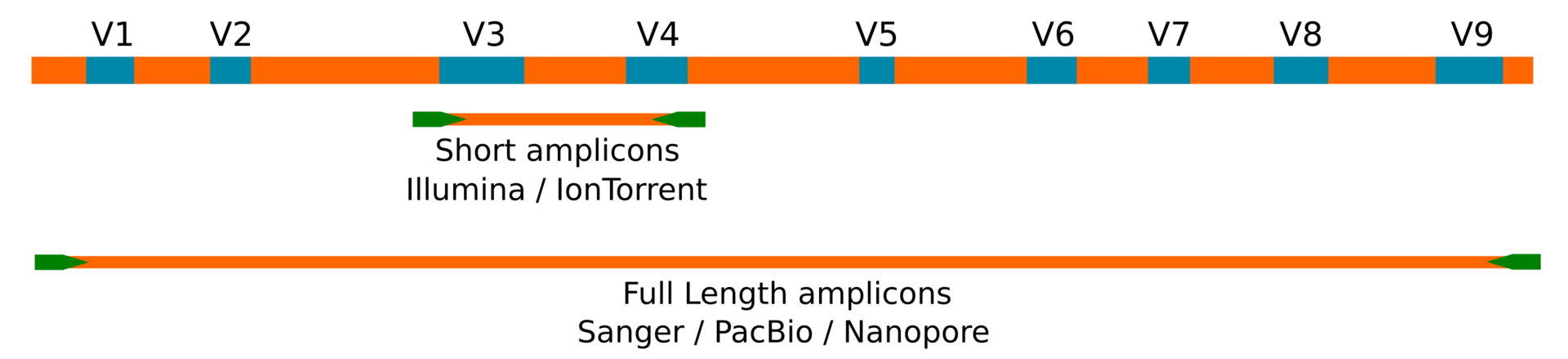
Los métodos de segunda generación Illumina e IonTorrent producen secuencias útiles de alrededor de ~500bp. Sanger, PacBio y Nanopore, permiten alcanzar el gen en toda su extensión con secuencias emparejadas o únicas de ~1500bp.
Secuenciación completa del gen ribosomal 16S (Full Length 16S)
En 2009, finalmente llegan los métodos de tercera generación de Pacific Bioiscience (PacBio) y Oxford Nanopore Technologies. Ambos nos prometen secuencias muy largas. Las dudas se quedan en los valores de calidad de las lecturas. Mientras que Nanopore, hasta la fecha, cubre un número importante de aplicaciones por la simplicidad en la preparación de librerías, la secuenciación, el bajo coste de las infraestructuras, la velocidad en la obtención de los datos y la portabilidad (perdonad si es poco), PacBio se impone por la calidad de los datos. Ambos métodos producen secuencias de longitudes que pueden superar las cien kilobases (100.000 nucleótidos), siendo hoy en día, elección obligada en la secuenciación de genomas completos. La baja calidad de Nanopore se corrige generalmente con la cobertura y/o secuenciaciones híbridas (Nanopore + Illumina).
PacBio entra ya directamente, con su protocolo de secuenciación HiFi, con secuencias de altísima calidad, cuya fiabilidad se sitúa por la mayoría alrededor de "Q50" con valores que pueden alcanzar "Q90", es decir, un error cada 100.000-1.000.000.000 nucleótidos. Esto lo hace apto para los análisis de descripción taxonómica donde necesitamos un alto grado de confianza para una correcta asignación taxonómica de cada una de las secuencias obtenidas. Lo que hace más interesante a PacBio para este tipo de estudios, es que finalmente podemos volver a secuenciar el gen ribosomal 16S completo y anotar hasta el rango de especie (o de clon), o hasta el operón ribosomal entero, a un coste por muestra parecido a los de segunda generación. Hemos vuelto a la época de la secuenciación del gen completo, pero con las ventajas de costes, calidad y tiempos de obtención de datos, típicos de los modernos métodos de secuenciación paralela.
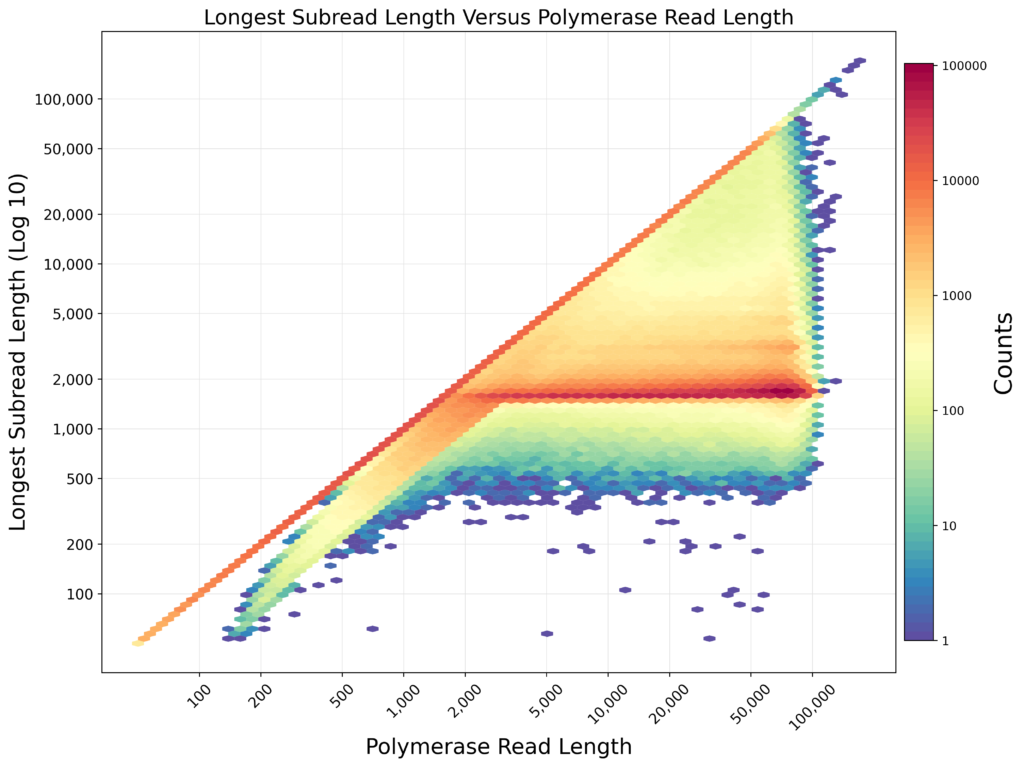
Representación de la calidad de la secuenciación por PacBio de los amplicones del gen ribosomal 16S. PacBio produce secuencias que se leen múltiples veces (eje X), mejorando la calidad. Las Subread representan las múltiples lecturas ya colapsada en una sola (eje Y). El mapa de colores representa el número de lecturas.
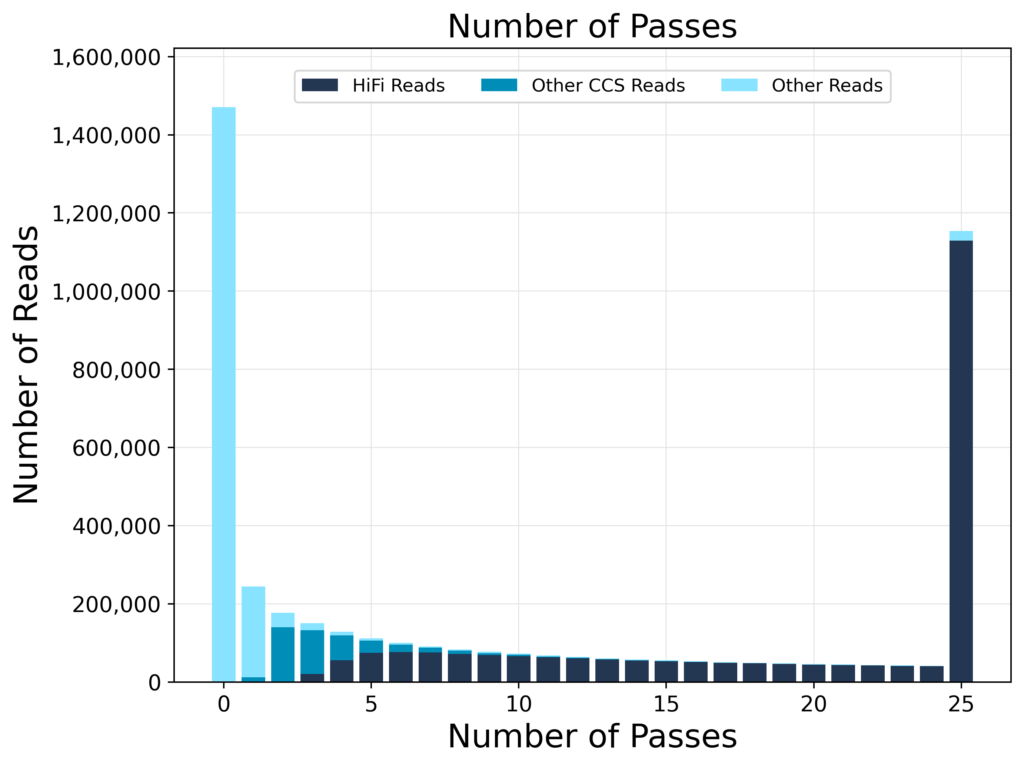
Cada secuencia de PacBio se lee múltiples veces mejorando la calidad. Las que superan un umbral de número de pasos se definen HiFi.
Ahora nos encontramos con 10 años de datos acumulados de secuencias parciales y hay que volver a "repoblar" las bases de datos de secuencias completas del gen ribosomal 16S. Al día de hoy, esta "falta de información" representa un problema que, poco a poco, se irá solventando con la aportación de información nueva, completa y fiable.
Más información sobre PacBio y sus aplicaciones en metagenómica en: PacBio BLOG